Start a new experiment by typing begin from the command line
(the begin
command can also be launched from the GUI pull-down menus by selecting File - New -
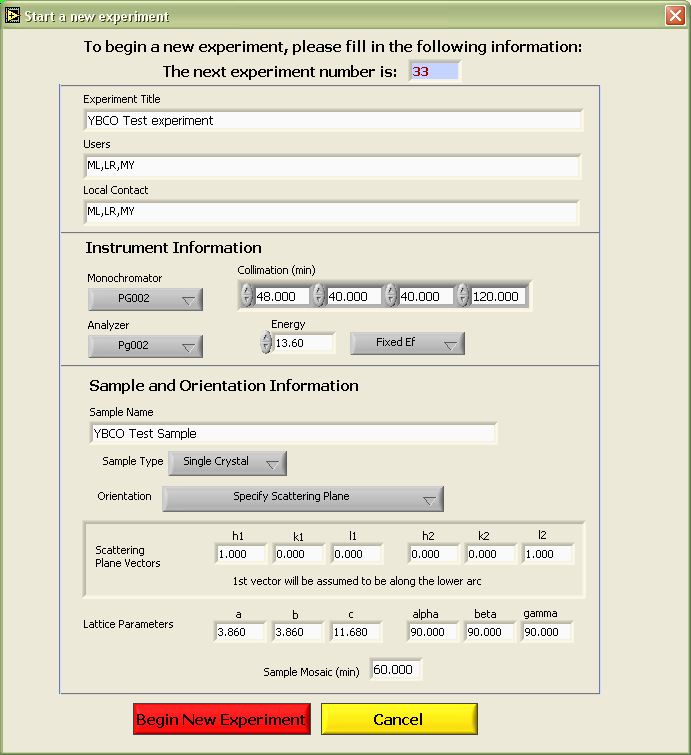
Experiment). This will launch a dialog window (see the
figure below) with several fields which must be filled in before the new
experiment can begin. The following elements must be supplied:
experiment title, users, local contact, monochromator/analyzer
selection, collimation, energy information (fixed Ei/Ef and the fixed
energy value) and sample information (name, type, lattice constants, and
alignment information (if applicable)). Use the tab key (or
the mouse) to switch between fields in this dialog window.
If a single crystal sample is selected, the user can choose how the
initial orientation will be specified. The available choices are:
1. Specify a scattering
plane - vectors which define the scattering plane and the lattice
constants must be given with this option. NOTE: with this option,
the 1st vector which defines the plane (h1,k1,l1) is assumed to be along
the lower arc. 2. Start with
no orientation specified - the lattice constants are specified
with this option but no moves in Q-space will be allowed until a valid
orientation has been specified. For information on how to define
an orientation see How to align a
single crystal. 3. Keep last
used orientation - this option will retain the orientation last
used in the program. This option is included primarily for cases
where the same sample will be used in sebsequent experiments.
After the form is filled out, hit the Begin
New Experiment button to initialize the new experiment.
2. Align
the monochromator
The first step in aligning the spectrometer is the alignment of the
monochromator crystal. NOTE:
this should ONLY be
performed under the supervision of the local contact. The
monochromator alignment is accomplished through the following set of
commands: defcount monitor preset time 2 scanrel m1 -1 1 .1
The above commands set the default counter to be the monitor, the
preset to 2 seconds and performs a relative scan of motor m1 +/- 1
degree in steps of 0.1 degrees. If the resulting scan is
symmetric, the m1 position can be driven to the center of mass by: com m1
If the scan is not symmetric but the nominal peak position is given by
the motor value PEAK, the m1 position can be driven to this value by: drive m1 PEAK
The monochromator should now be aligned but the default counting
channel should be set from the monitor back to the detector: defcount detector
3. Calibration
using powder standard
(a)
The next steps involve running a standard sample to calibrate the
energy of the incident beam (zero of motor m2) and the zero of sample
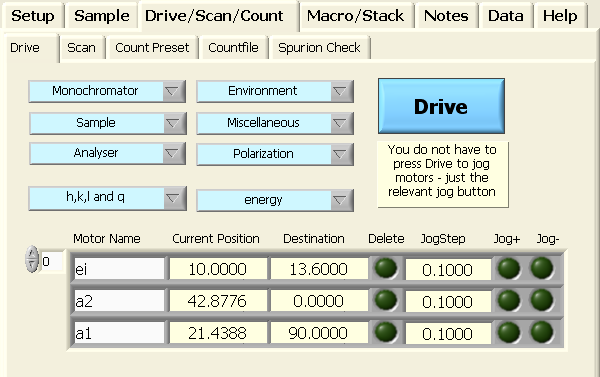
scattering angle s2. First, drive the monochromator
to the selected energy value (13.6 meV in this example), the analyzer scattering
angle (s2) to the straight-through position, and the analyzer orientation
angle (a1) to a transmission setting (Motors > Drive)
This command can also be issued from the GUI by making the selections
shown in the figure below and hitting the blue Drive
button:
(b)Cursory check of the zero ofs2. NOTE: This step may be skipped
if the alignment is believed to be fairly accurate as it will only set a
rough value for the s2 zero angle. It is suggested that an Al-B attenuator be used to
prevent saturation of the detector. Scan s2 through the
direct beam, from –1 to 1 in steps 0f 0.1 for a preset time of 1 second.
This rough alignment is performed to ensure the ranges for the standard
sample calibration are adequate. As mentioned above, if the
alignment is believed to be fairly accurate, this step can be skipped.
(c) Nickel powder calibration of energy and
zeroes.
Put the Ni powder can at the sample position and remove all PG filters
(The filter removal is necessary as the calibration uses lambda/2
reflections).
The standard powder calibration is designed to be fully automated and
should be run from the GUI. Details of the procedure are available
in the help document for the calibrate command.
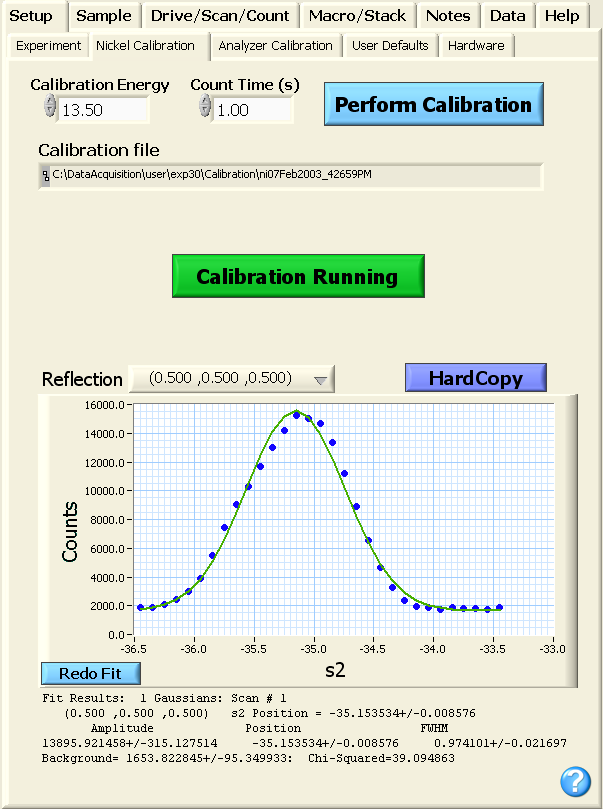
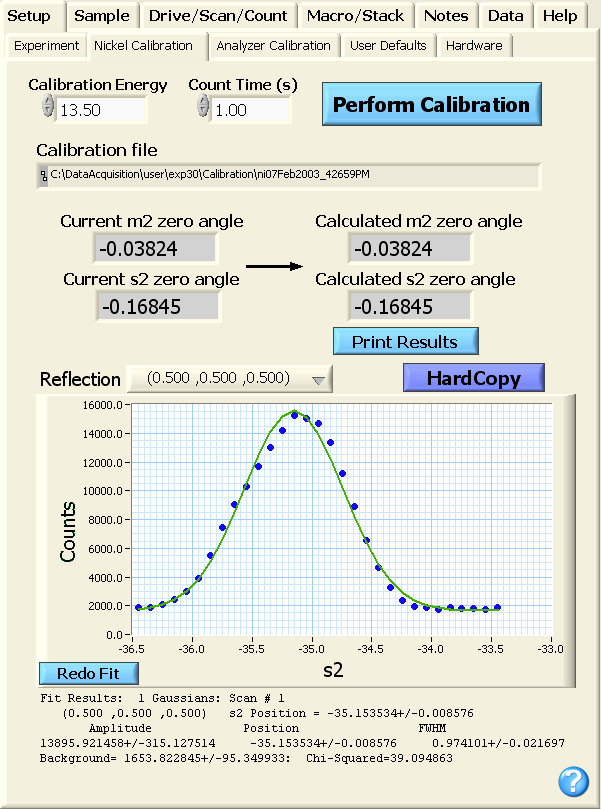
The calibration is initiated from the GUI by selecting the Nickel Calibration
tab from the Setup
top-level tab. To begin a calibration, the user types in the energy at
which the Ni calibration will be performed (13.5 meV in the example
shown below), selects an appropriate count time
(unless collimation is fairly tight, a count time of 1 second should be
adequate), and clicks the blue Perform Calibration
button. While the calculation is taking place, a green Calibration Running button
is visible as shown in the image below.
During the calibration, the user can examine the scans and the
generated fit by selecting the Reflection
pull-down menu. If any of the fits are not satisfactory, the Redo Fit
button will bring up a popup box where initial guess parameters can be
input. Following the completion of the calibration, the Calibration Running
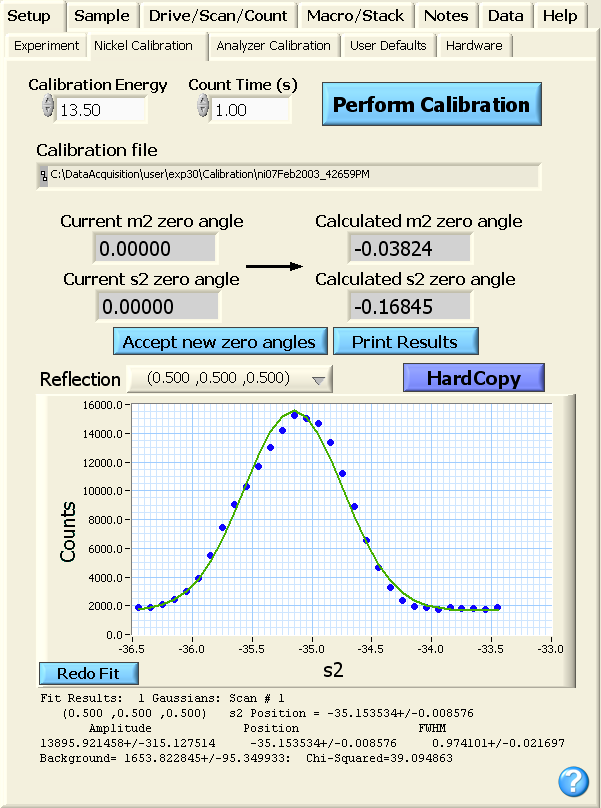
button will be replaced by the boxes shown below where the current and calculated zero
angles for motors m2 and s2 are shown. The user can examine
the output in the logfile or can click on the blue Print Results
button to produce a hardcopy summarizing the fit information from the
calibration.
If the calibration seems satisfactory, the uses accepts the new zero
angles by clicking on the blue Accept new zero
angles button.
After accepting the new zero angles, the Accept new zero
angles button will disappear and the current and calculated zero
angles should now be identical as
shown in the bottom image. This will complete the Ni powder
calibration. A hardcopy of all the scans and the individual fits
can be produced by hitting the blue HardCopy
button.
4.Analyzer alignment
A strong incoherent scatterer should be placed at the sample position.
The typical options are to use the same Ni powder used above
(being sure to be well away from any powder lines) OR use a vanadium
sample. The analyzer is optimized by scanning some combination of a1, a2,ef ande. As
most neutron scatterers seem to have their own favorite series of scans
for completing this process (and because analyzer scans are not always
symmetric) the alignment procedure for the analyzer has not been
automated. Once a1 and a2
have been optimized (and they are positioned at that optimal position),
the process of setting the zero angles has been automated and can be
accessed from the GUI by selecting the Analyzer Calibration
tab from the Setup
top-level tab. The zero angles are set by hitting the blue Make
current analyzer setting elastic button.
The spectrometer is now aligned and the next step in setting up an
experiment involves aligning the sample.